analysis from R. Basom (from the Hutch):
I've attached your results, which were generated via the following method, using the Bioconductor package, "Ringo":
* Read NimbleGen pair files into R
* Remove control probes
* Tukey biweight mean center log2 ratios within each array (all ratios were formulated such that the estrogen treated sample is always in the numerator)
* Smooth log2 ratio values by computing a running median, using a window size of 1000 bases
* Within each array, use the log2 ratio distribution to calculate the threshold to be used for peak detection, then detect peaks such that at least 3 adjacent probes must be above threshold.
* Identify the overlap between A02 and A03, excluding A01 using the Bioconductor package "ChIPpeakAnno." There are 96 regions from A02 that overlap with 99 regions from A03 - but not A01.
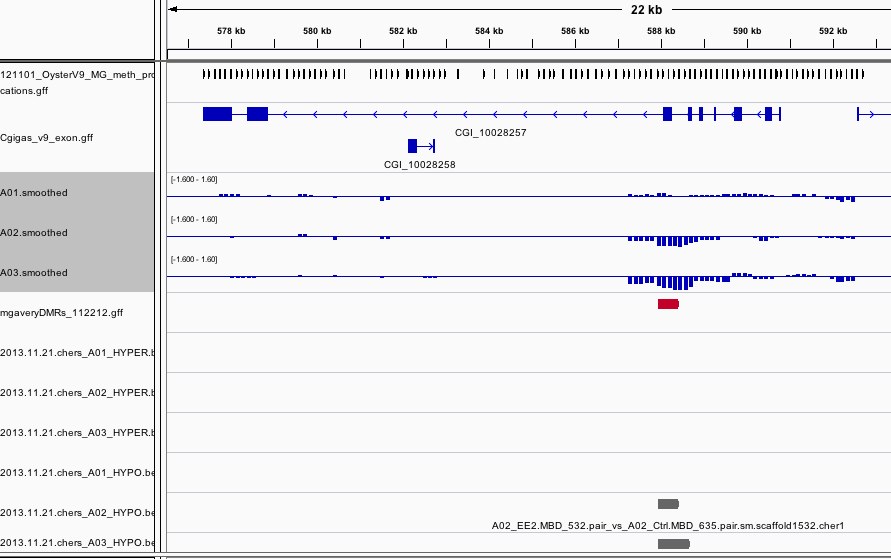
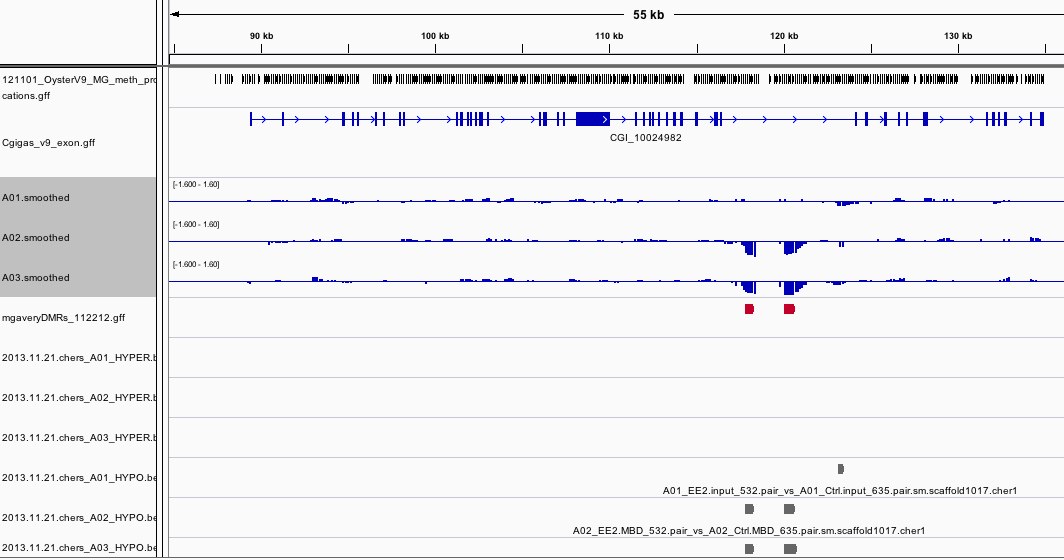
The results can be viewed in IGV. If you don't already have an IGV genome configured for the Pacific Oyster, there's an IGVgenome subdirectory that contains one, though the oyster fasta file will have to be placed in this same directory (there are instructions in the README). There's also a subdirectory of genomeBrowserTracks, that has wig files with the smoothed values from each array, as well as bed files with the peaks that were detected for A01, as well as files that have peaks that were detected in both A02 and A03, but not A01. I've loaded the genome browser tracks into IGV and have done some configuration to assist with visualization. If you'd like to use the configuration I've put together, you can load the session file that's attached in the results. There's a screen shot to provide an example of how I've configured the tracks in IGV. There's also an Excel file that has the 96 regions from A02 that overlap with A03, but not A01. If you already have IGV open, this file has a column of hyperlinks that can be clicked on to direct IGV to each overlap region.
Hypermethylated Regions:
Analysis with mRNA hits
91 regions overlapped w/ mRNA
genefish:bin Mackenzie$ ./intersectBed -a /Volumes/web-1/bivalvia/wholegenomefiles_MBDbsSeq_gill/gffs/oyster_v9_glean_final_rename_mRNA.gff -b /Volumes/web-1/bivalvia/array/2013.09.27.mgavery/2013.09.27.A03overlapWithA02.bed > /Volumes/web-1/bivalvia/array/arrayRegions_mRNA.txt
genefish:bin Mackenzie$ wc /Volumes/web-1/bivalvia/array/arrayRegions_mRNA.txt
91 819 6013 /Volumes/web-1/bivalvia/array/arrayRegions_mRNA.txt
Pulled the 91 regions into galaxy and imported fasta of oyster genes (oyster_v9_gene.fasta)
Did a fasta to tabular, joined on the gene ID, cut the gene ID and sequences only to a new file and converted tabular to fasta.
SAME ANALYSIS as above BUT W/ all gene hits (i.e. mRNA)
82 SPIDs put in, given 76 DAVID IDs
ALL OYSTER GENES ARE USED AS BACKGROUND ~~SEE BELOW FOR ONLY GENES ON THE ARRAY AS BACKGROUND
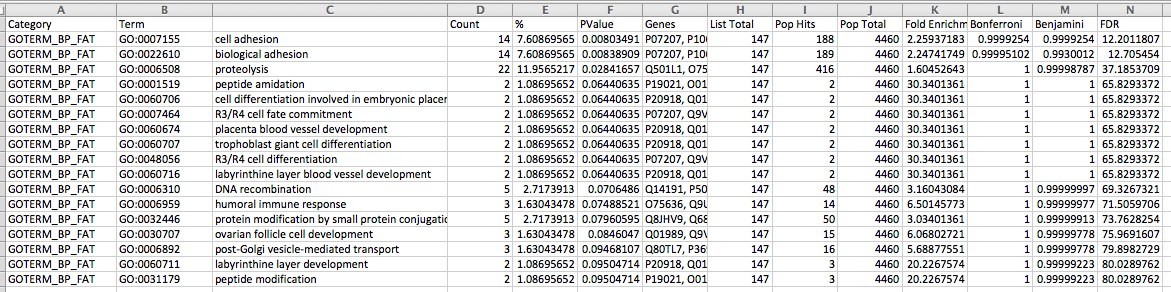
snapshot
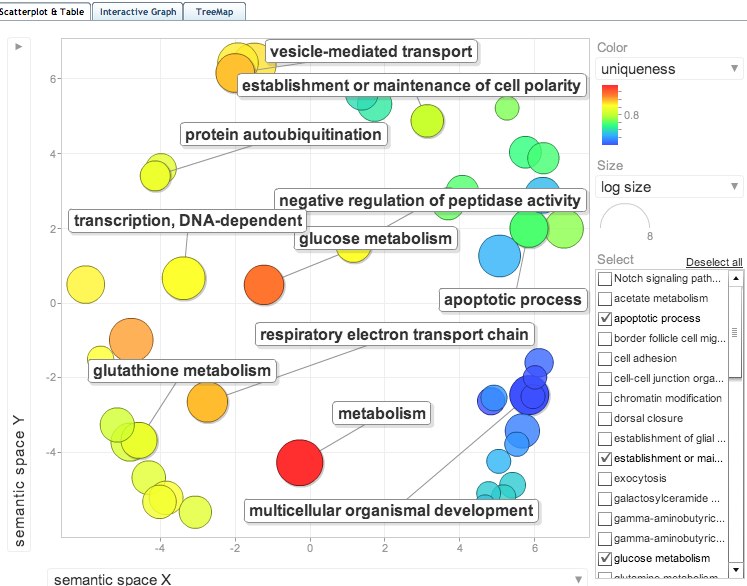
joined the SP_accessions to GO terms in SQLshare and put GO terms into ReviGO. Visualization below:
Not many differences from above (Check significance after correction, none are below 0.05) Only addition is regulation of neurotransmitter levels. The EXACT SAME 9 SPIDs contributed to the enriched categories so the ReviGO plots above would be the same.
Hypomethylated Regions:
As it turns out the above 96 regions are hypermethylated in the EE2 treated sample, there are another 287 regions that are hypomethylated in the EE2 treated.
When joined to mRNA 268 regions overlapped with mRNA (these may not be unique regions of the 287 since mRNAs also overlap)
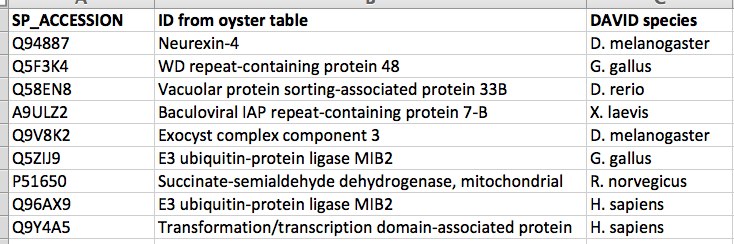
I identified the genes that overlapped with these hypomethylated regions and annotated them to their SPIDs. There were 201 unique SPIDs in this list. I used those as the gene list and all genes on the array as background for enrichment analysis in DAVID
Here is the output of enriched processes:
Here are the enriched gene IDs:
11/01/13
I am interested in seeing if the differentially methylated regions are primarily in exons or introns or if they cross an intron exon boundary:
Initial summary:
96 hypermethylated regions (compared to control): 90 overlapped with mRNA
287 hypomethylated regions : 256 overlapped with mRNA
~~it is not surprising that this may overlapped with genes since the tiling array was designed to cover genes (+2kb upstream)~~
How many regions overlapped with intron/exon boundaries??
First, designed a gff to span 10 bp upstream from an intron (aka 10 bp upstream of an exon intron boundary):
bedtools code:
genefish:bin Mackenzie$ ./flankBed -i /Volumes/web-1/trilobite/Crassostrea_gigas_v9_tracks/Cgigas_v9_intron.gff -g /Volumes/web-1/cnidarian/qDOD_scaffold_length.txt -l 10 -r 0 > /Volumes/web-1/bivalvia/array/flankIntron.txt
Then, performed an intersect bed to count the number of times a differentially methylated region intersected one of these boundaries
Summary:
Of the 90 hypermethylated regions overlapping with a gene 47 (or 52%) crossed an intron/exon boundary (a majority only crossed one boundary, but up to 6 boundaries were crossed by a single region)
Of the 256 hypomethylated regions overlapping with a gene 122 (or 48%) overlapped 1 or more boundaries (similar to hypermethylated regions, a majority only crossed one boundary, but up to 6 boundaries were crossed by a single region)
How many differentially methylation regions overlap ONLY with coding regions?
I pulled out the subset of regions that did overlap with mRNA, but did NOT overlap with an exon/intron boundary and performed an intersect bed to count how many regions overlapped with just an exon (and can also draw conclusions that those that do not overlap only overlap with an intron).
hypermethylated: there were 43 regions that overlapped with mRNA, but did NOT cross an intron/exon boundary. I did 2 intersectBed with these 43 regions - 1 with the exon.gff 1 with the intron.gff. What I found was the 32 overlapped with an intron only 6 overlapped with an exon only and an additional 5 came up as intersecting with both an intron and an exon (this probably has something to do with the gff I created to identify boundaries (only 10bp upstream from intron).
But! to summarize: 96 regions: 90 are in genes (52 cross exon/intron boundary another 32 are in introns only and 6 are just in exons), 6 are outside of gene
hypomethylated:
there were 134 regions that overlapped with mRNA, but did NOT cross an intron/exon boundary. I did 2 intersectBed with these 134 regions - 1 with the exon.gff 1 with the intron.gff. What I found was the 114 overlapped with an intron only 4 overlapped with an exon only and an additional 16 came up as intersecting with both an intron and an exon (this probably has something to do with the gff I created to identify boundaries (only 10bp upstream from intron).
But! to summarize: 287 regions: 256 are in genes (138 cross exon/intron boundary another 114 are in introns only and 4 are just in exons), 31 are outside of gene
files I worked with:
intron and exon canonical files in trilobite
intron/exon boundary gff I generated (described above)
generated bed files with
How big are these regions?
min: 231 bp
max: 7,979 bp
average: 983 bp
Determine how to validate these?
-Is there a way to pick the 'best' regions to validate? (log2s?)
-How much would it cost to do pyrosequencing of these regions?
How many CG sites per regions?
Did some intersect beds
1. count the number of CG per DMR
genefish:bin Mackenzie$ ./intersectBed -c -a /Volumes/web-2/bivalvia/array/2013.09.27.mgavery/2013.09.27.A03overlapWithA02.bed -b /Volumes/web-2/trilobite/Crassostrea_gigas_v9_tracks/Cgigas_v9_CG.gff >/Users/Mackenzie/Desktop/hyperarray_CG.txt
2. overlap DMR with mRNAs using the -wb option to get gene id for each DMR
genefish:bin Mackenzie$ ./intersectBed -wb -a /Volumes/web-2/bivalvia/array/2013.09.27.mgavery/2013.09.27.A03overlapWithA02.bed -b /Volumes/web-2/trilobite/Crassostrea_gigas_v9_tracks/Cgigas_v9_gene.gff >/Users/Mackenzie/Desktop/hyperarray_mRNA.txt
Interestingly, 3 of the regions did not contain a CG. Why were they identified as being differentially methylated?
;%20Advanced%20Biomedical%20Computing%20Center%20(ABCC).jpg)


;%20Science%20Applications%20International%20Corporation%20(SAIC)-1.jpg)